និយមន័យ
Amyotrophies ឆ្អឹងខ្នងគឺជាក្រុមនៃជំងឺតំណពូជដែលបណ្តាលមកពីដំណើរការ dystrophic បឋមនៅក្នុងស្នែងផ្នែកខាងមុខនៃខួរឆ្អឹងខ្នងដែលត្រូវបានបង្ហាញដោយ paresis flaccid និងសាច់ដុំ atrophy ។
ពួកវាបង្កើតបានប្រហែល 7% នៃជម្ងឺណឺរ៉ូនម៉ូទ័រផ្សេងទៀត ដែលរួមបញ្ចូលផងដែរនូវជំងឺក្រិនសរសៃឈាមអាមីតូត្រូហ្វីក ជម្ងឺក្រិនសរសៃឈាមបឋមដ៏កម្រ និងជំងឺដាច់សរសៃឈាមខួរក្បាល។ អត្រាប្រេវ៉ាឡង់នៃ amyotrophy ឆ្អឹងខ្នងនៅក្នុងប្រជាជនគឺពី 0.65 ទៅ 1.6 ក្នុង 100,000 ប្រជាជន។
ចំណាត់ថ្នាក់
ទម្រង់ព្យាបាលផ្សេងៗនៃ amyotrophy ឆ្អឹងខ្នងមានភាពខុសគ្នាទៅតាមអាយុនៃការចាប់ផ្តើម អត្រានៃការវិវត្តន៍ និងប្រភេទនៃមរតកនៃជំងឺ។
ទម្រង់ទូទៅបំផុត៖
I. Amyotrophies ឆ្អឹងខ្នងនៃកុមារភាព និងវ័យជំទង់៖
1) អាមីតូត្រូហ្វីលឆ្អឹងខ្នងរបស់ទារកដែលមានសាហាវស្រួចស្រាវ (Werdnig-Hoffmann);
2) amyotrophy ឆ្អឹងខ្នងទារករ៉ាំរ៉ៃ;
3) amyotrophy ឆ្អឹងខ្នងអនីតិជន (Kugelberg-Welander) ។
II. Amyotrophy ឆ្អឹងខ្នងចំពោះមនុស្សពេញវ័យ៖
1) bulbospinal amyotrophy (Kennedy);
2) amyotrophy ឆ្អឹងខ្នង distal (Duchenne-Arana);
3) scapulo-peroneal amyotrophy (Vulpiana) ។
គ្លីនិក និងលក្ខណៈវិនិច្ឆ័យរោគ
1. លក្ខណៈគ្លីនិកទូទៅ៖ ភាពទន់ខ្សោយស៊ីមេទ្រីនៃសាច់ដុំជិត មិនសូវជាញឹកញាប់។ Asymmetry នៃការខូចខាតដល់សាច់ដុំនៃចុងបំផុត និងការជាប់ពាក់ព័ន្ធនៃក្រុមសាច់ដុំ bulbar គឺកម្រមានណាស់។ តាមក្បួនមួយ មិនមានរោគសតិអារម្មណ៍ទេ;
2. លក្ខណៈវិនិច្ឆ័យរោគវិនិច្ឆ័យ៖
- លក្ខណៈតំណពូជនៃជំងឺ (ប្រភេទនៃមរតកមិនតែងតែងាយស្រួលក្នុងការបង្កើតទេ);
- សាច់ដុំ atrophy ជាមួយ fasciculations, fibrillations;
- EMG - រូបភាពនៃការខូចខាតដល់ស្នែងផ្នែកខាងមុខនៃខួរឆ្អឹងខ្នង;
- អវត្ដមាននៃជំងឺសរសៃប្រសាទនិងអាងត្រគាក;
- វគ្គសិក្សារីកចម្រើន;
- fascicular atrophy នៃសរសៃសាច់ដុំនៅលើការធ្វើកោសល្យវិច័យ។
3. លក្ខណៈគ្លីនិកនៃទម្រង់បុគ្គល៖
1) Werdnig-Hoffmann spinal amyotrophy (malignant infantile spinal amyotrophy) គឺជាជំងឺ autosomal recessive, ហ្សែន mutant ត្រូវបានគូសផែនទីទៅនឹងក្រូម៉ូសូមទី 5 ។ ហ្សែនមួយទៀតត្រូវបានគេកំណត់អត្តសញ្ញាណដែលផ្តល់នូវការទប់ស្កាត់ apoptosis - ការស្លាប់តាមកម្មវិធីនៃសរសៃប្រសាទ។ វាគឺជាហ្សែននេះដែលជារឿយៗអវត្តមានចំពោះអ្នកជំងឺធ្ងន់ធ្ងរ។ ឧប្បត្តិហេតុនៃជំងឺនេះគឺ 1: 25,000 ទារកទើបនឹងកើត។ ថ្មីៗនេះវាត្រូវបានបែងចែកទៅជាស្រួចស្រាវ (តាមពិតទម្រង់ Werdnig-Hoffman) និងអាមីតូត្រូហ្វីលឆ្អឹងខ្នងរ៉ាំរ៉ៃ។
ទម្រង់ស្រួចស្រាវបង្ហាញរាងដោយខ្លួនឯងក្នុងរយៈពេល 5 ខែដំបូងនៃជីវិតហើយបញ្ចប់យ៉ាងធ្ងន់ធ្ងរក្នុងរយៈពេល 1,5 ឆ្នាំ។ នៅក្នុងទម្រង់រ៉ាំរ៉ៃមានទម្រង់កុមារភាពដំបូង (ចាប់ផ្តើមមុនអាយុ 1.5-2 ឆ្នាំ ស្លាប់ដោយ 4-5 ឆ្នាំពីការបរាជ័យផ្លូវដង្ហើម ជំងឺរលាកសួត) និងទម្រង់យឺត (ចាប់ផ្តើមមុន 2 ឆ្នាំ អចល័តនៅអាយុ 10 ឆ្នាំ ស្លាប់នៅអាយុ ១៥-១៨ ឆ្នាំ) ។ រោគសញ្ញាសំខាន់ៗ៖ paresis នៃជើងជិតៗ បន្ទាប់មកដៃ សាច់ដុំប្រម៉ោយ សាច់ដុំផ្លូវដង្ហើម មានភាពបត់បែន សរសៃពួរ សាច់ដុំគ្រោងឆ្អឹង និងអណ្តាត កន្ត្រាក់ ខូចទ្រង់ទ្រាយឆ្អឹង រោគសញ្ញានៃដុំពក ជម្ងឺ hyperhidrosis ទូទៅ។ EMG បង្ហាញពីសកម្មភាពជីវអេឡិចត្រិចដោយឯកឯងនៅពេលសម្រាកជាមួយនឹងវត្តមាននៃសក្តានុពល fasciculation ។ ក្នុងអំឡុងពេលនៃការកន្ត្រាក់ដោយស្ម័គ្រចិត្ត ការកាត់បន្ថយសកម្មភាពអគ្គិសនីជាមួយនឹងចង្វាក់ "របងជ្រើសរើស" ត្រូវបានកត់ត្រា។ សកម្មភាពរបស់អង់ស៊ីមសេរ៉ូមមិនផ្លាស់ប្តូរទេ។ ការពិនិត្យរោគវិទ្យាបានបង្ហាញពីការថយចុះនៃចំនួនកោសិកានៅក្នុងស្នែងខាងមុខនៃខួរឆ្អឹងខ្នង នៅក្នុងស្នូលម៉ូទ័រនៃដើមខួរក្បាល និងការផ្លាស់ប្តូរ degenerative នៅក្នុងពួកវា។ នៅក្នុងសាច់ដុំ - fascicular atrophy នៃសរសៃសាច់ដុំ;
2) Kugelberg-Welander amyotrophy ឆ្អឹងខ្នង (ទម្រង់អនីតិជនឬ pseudomyopathic) - ជំងឺដែលមានប្រភេទ autosomal recessive នៃមរតក។ ប្រភេទនៃវគ្គសិក្សា និងការចែកចាយនៃសាច់ដុំ atrophies គឺស្រដៀងទៅនឹងជំងឺសាច់ដុំ Erb-Roth ប៉ុន្តែមានការរីករាលដាលនៃសាច់ដុំ។ EMG បញ្ជាក់ពីលក្ខណៈឆ្អឹងខ្នងនៃសាច់ដុំ atrophy ។ រោគសាស្ត្រនៃសាច់ដុំកំឡុងពេលធ្វើកោសល្យវិច័យ រួមជាមួយនឹងជំងឺសរសៃប្រសាទ (fascicular) amyotrophy បង្ហាញពីសញ្ញានៃការខូចខាតសាច់ដុំបឋម (សាយភាយ) ។
ជំងឺនេះចាប់ផ្តើមពីអាយុពី 2 ទៅ 15 ឆ្នាំ ហើយវិវត្តយឺតណាស់។ ភាពទន់ខ្សោយនៃសាច់ដុំ និងស្ពឹករីកដុះដាលដំបូងនៅជើងជិតៗ ខ្សែក្រវាត់អាងត្រគាក ហើយរាលដាលបន្តិចម្តងៗទៅសាច់ដុំនៃខ្សែស្មា។ អ្នកជំងឺខ្លះមាន pseudohypertrophy នៃសាច់ដុំ, hyperfermentemia (ជាពិសេសការកើនឡើងនៃ CPK) ដែលនាំទម្រង់នេះខិតទៅជិត PMD ។ មិនមានការខូចទ្រង់ទ្រាយឆ្អឹងឬការដកសាច់ដុំទេ។ ភាពមិនប្រក្រតីនៃចលនា Bulbar កើតឡើងនៅដំណាក់កាលចុងក្រោយនៃជំងឺ។ អ្នកជំងឺរក្សាសមត្ថភាពក្នុងការថែទាំខ្លួនឯងបានយូរ ហើយជារឿយៗអាចធ្វើការបានច្រើនឆ្នាំ។
3) Kennedy bulbospinal amyotrophy - ជាជំងឺដែលទាក់ទងនឹង X-linked recessive disease ដែលជាធម្មតាបង្ហាញខ្លួនវាបន្ទាប់ពី 30 ឆ្នាំ។ វាត្រូវបានបង្កឡើងដោយការផ្លាស់ប្តូរជាក់លាក់មួយនៅក្នុងហ្សែន receptor androgen ដែលបានគូសផែនទីនៅលើក្រូម៉ូសូម X ។ មានតែបុរសទេដែលឈឺ។ វាចាប់ផ្តើមជាមួយនឹងផ្នែកជិតៗនៃអវយវៈបន្ទាប់ពី 10-20 ឆ្នាំ (ជួនកាលមុននេះ) ជំងឺ bulbar លេចឡើងក្នុងទម្រង់នៃការ atrophy និងភាពទន់ខ្សោយនៃសាច់ដុំ masticatory, dysphagia និង dysarthria ។ ដោយសារតែការវិវត្តន៍យឺតយ៉ាវនៃជំងឺនេះ ជំងឺ bulbar មិននាំឱ្យមានការចុះខ្សោយធ្ងន់ធ្ងរនៃមុខងារសំខាន់ៗអស់រយៈពេលជាច្រើនឆ្នាំ។ មានការញ័រដៃ និងក្បាល រំឭកពីការញ័រសំខាន់ៗ។ រោគសញ្ញាលក្ខណៈមួយគឺការកន្ត្រាក់នៃសាច់ដុំផ្នែកខាងក្នុង និងអណ្តាត និងបញ្ហាប្រព័ន្ធ endocrine (រោគស្ត្រី, ការថយចុះកម្លាំង, ពងស្វាស, ជំងឺទឹកនោមផ្អែម) ។ វគ្គសិក្សាគឺយឺត, ការព្យាករណ៍សង្គមជាទូទៅអំណោយផល;
4) amyotrophy ឆ្អឹងខ្នង distal របស់ Duchenne-Aran ។ របៀបនៃការទទួលមរតកគឺ autosomal dominant ឬ autosomal recessive ករណីកម្រកើតមានជាញឹកញាប់។ ចាប់ផ្តើមបន្ទាប់ពី 20 ឆ្នាំ (ជាធម្មតានៅ 30-50) ។ ទម្រង់បុរាណនៃជំងឺនេះត្រូវបានកំណត់លក្ខណៈដោយការចាប់ផ្តើមនៅផ្នែកចុងបំផុតនៃចុងខាងលើ ("ដៃក្រញ៉ាំ") ក្រោយមកការដាច់រលាត់រាលដាលដល់កំភួនដៃ ស្មា ("ដៃគ្រោងឆ្អឹង") ហើយបន្ទាប់ពីជាច្រើនឆ្នាំ ភាពទន់ខ្សោយនៃសាច់ដុំ។ ក្រុម peroneal និងសាច់ដុំផ្សេងទៀតនៃជើងទាប ភ្លៅ និងដងខ្លួនកើតឡើង។ រោគសញ្ញាពីរ៉ាមីតស្រាលកើតឡើង។ នៅដំណាក់កាលដំបូងនៃដំណើរការអាចមានដំបៅមួយចំហៀង monoparesis (Mozolevsky Yu. V. et al ។ , 1988) ។ ការរួមផ្សំជាមួយនឹងជំងឺផាកឃីនសុន ឌីស្តូនៀ រមួលក្រពើ និង myoclonic hyperkinesis គឺអាចធ្វើទៅបាន (Makarov A. Yu., 1967) ។ ជំងឺនេះកម្រជះឥទ្ធិពលយ៉ាងខ្លាំងដល់ស្ថានភាពសង្គមរបស់អ្នកជំងឺដោយសារតែការវិវត្តន៍យឺតយ៉ាវខ្លាំង (លើកលែងតែករណីដែលមានរោគសាស្ត្ររួមគ្នា);
5) ទម្រង់ Vulpian scapulo-peroneal ។ វាចាប់ផ្តើមនៅចន្លោះពី 20-40 ឆ្នាំជាមួយនឹងការចុះខ្សោយនៃសាច់ដុំនៃខ្សែស្មា (ចលនាមានកំណត់នៅក្នុងសន្លាក់ស្មា "រាងស្លាប" ស្មា យូរ ៗ ទៅភាពទន់ខ្សោយនៃផ្នែកបន្ថែមនៃជើងនិងជើងត្រូវបានបន្ថែមទោះបីជា លំដាប់បញ្ច្រាសគឺអាចធ្វើទៅបាន។ ការធ្វើរោគវិនិច្ឆ័យឌីផេរ៉ង់ស្យែលគួរតែត្រូវបានធ្វើឡើងជាចម្បងជាមួយនឹងជំងឺ myodystrophy scapuloperoieal របស់ Davidenkov ។ ការវិវត្តន៍មានភាពយឺតយ៉ាវ ជារឿយៗអ្នកជំងឺវ័យចំណាស់ដែលមានជំងឺពី 30-40 ឆ្នាំអាចផ្លាស់ទីដោយឯករាជ្យ។
4. ទិន្នន័យស្រាវជ្រាវបន្ថែម៖
- EMG, ENMG ។ ជាមួយនឹង EMG សរុប សញ្ញានៃដំបៅស្នែងខាងមុខ៖ នៅក្នុងរបៀបសម្រាក សកម្មភាពជីវអគ្គិសនីដោយឯកឯងក្នុងទម្រង់នៃសក្ដានុពលនៃសារពើពន្ធដែលមានទំហំរហូតដល់ 200 μV ជាមួយនឹងការកន្ត្រាក់ដោយស្ម័គ្រចិត្ត EMG ថយចុះជាមួយនឹងចង្វាក់ "របងជ្រើសរើស" និង ហូរចេញយ៉ាងខ្លាំងនៅក្នុងការខូចខាតសាច់ដុំធ្ងន់ធ្ងរ។ ENMG បង្ហាញពីការថយចុះនៃទំហំនៃការឆ្លើយតប M និងចំនួននៃម៉ូទ័រដំណើរការ។ ល្បឿននៃការដំណើរការនៃកម្លាំងរុញច្រានប្រែប្រួលអាស្រ័យលើទម្រង់នៃ amyotrophy ឆ្អឹងខ្នងនិងអាយុរបស់អ្នកជំងឺ;
- ការធ្វើកោសល្យវិច័យសាច់ដុំ (ការដាច់រលាត់ជាបណ្តុំគឺមានលក្ខណៈធម្មតា ផ្ទុយពីការសាយភាយ atrophy នៅក្នុង PMD);
- MRI ។ ពេលខ្លះវាជួយក្នុងការរកឃើញការ atrophy ខួរឆ្អឹងខ្នង;
ការសិក្សាអំពីសកម្មភាពរបស់ CPK, LDH, ALT នៅក្នុងសេរ៉ូមឈាម (ផ្ទុយទៅនឹង PMD វាធម្មតាឬកើនឡើងបន្តិច);
- ការប្រឹក្សាផ្នែកវេជ្ជសាស្ត្រ និងហ្សែន។
ការធ្វើរោគវិនិច្ឆ័យឌីផេរ៉ង់ស្យែល៖ 1. ជាមួយនឹង PMD ដែលថតចម្លង amyotrophy ឆ្អឹងខ្នង (ជំងឺ Erb-Roth, Davidenkov's scapuloperoneal myodystrophy ជាដើម)។
2. ការវះកាត់ឆ្អឹងខ្នងស្រួចស្រាវនៃ Werdnig-Hoffmann - ជាមួយនឹងមូលហេតុផ្សេងទៀតនៃរោគសញ្ញា "កុមារមិនប្រក្រតី" (ទម្រង់អាតូនិចនៃជំងឺខួរក្បាល ជំងឺ myopathy ពីកំណើត រោគសញ្ញា Marfan ជាដើម) ។
3. ជាមួយនឹងជំងឺក្រិនសរសៃឈាមអាមីតូត្រូហ្វីកនៅពេលក្រោយ - Kennedy bulbospinal amyotrophy ទម្រង់ផ្សេងទៀតនៃ amyotrophy ឆ្អឹងខ្នងចំពោះមនុស្សពេញវ័យ (ការធ្វើរោគវិនិច្ឆ័យចុងក្រោយជាញឹកញាប់ត្រូវបានធ្វើឡើងបន្ទាប់ពី 1-3 ឆ្នាំនៃការសង្កេតរបស់អ្នកជំងឺ) ។
4. ជាមួយនឹងជំងឺ myelopathy ischemic cervical (ឆ្អឹងខ្នង amyotrophy ចំពោះមនុស្សពេញវ័យ) ។
5. ជាមួយនឹងទម្រង់រ៉ាំរ៉ៃនៃជំងឺរលាកខួរក្បាលដែលកើតដោយធីក, ជំងឺ Lyme ។
6. ជាមួយនឹងរោគសញ្ញាក្រោយជំងឺខួរឆ្អឹងខ្នង។
វគ្គសិក្សានិងការព្យាករណ៍
អាស្រ័យលើអាយុនៃការចាប់ផ្តើមនៃជំងឺ។ ក្នុងទម្រង់ជាកុមារភាព លទ្ធផលគឺស្លាប់សូម្បីតែក្នុងករណីមានវគ្គរ៉ាំរ៉ៃ និងការចាប់ផ្តើមនៃជំងឺមុនអាយុ 2 ឆ្នាំ។ ក្នុងករណីទារក Kugelberg-Welander amyotrophy សមត្ថភាពក្នុងការថែទាំខ្លួនឯងនៅតែមានរយៈពេលជាច្រើនឆ្នាំ។ នៅក្នុងទម្រង់ផ្សេងទៀតនៃ amyotrophy ឆ្អឹងខ្នងចំពោះមនុស្សពេញវ័យ អាយុកាលជាមធ្យមមិនផ្លាស់ប្តូរទេ ប៉ុន្តែការកើនឡើងនៃកង្វះម៉ូទ័រជាធម្មតានាំឱ្យសកម្មភាពជីវិតមានកម្រិត និងសមត្ថភាពក្នុងការធ្វើការ (ច្រើនឆ្នាំបន្ទាប់ពីការចាប់ផ្តើមនៃជំងឺ) ។
គោលការណ៍នៃការព្យាបាល
ការសម្រាកនៅមន្ទីរពេទ្យសម្រាប់ការធ្វើរោគវិនិច្ឆ័យដំបូង វគ្គនៃការព្យាបាលថែទាំម្តងហើយម្តងទៀត (1-2 ដងក្នុងមួយឆ្នាំ) ក្នុងករណីមានជម្ងឺ decompensation បន្ទាប់ពីរបួស ជំងឺឆ្លង។ល។
ការព្យាបាលគឺផ្អែកលើគោលការណ៍ដូចគ្នានឹងជំងឺសាច់ដុំ - ធ្វើអោយប្រសើរឡើងនូវដំណើរការនៃសរសៃប្រសាទ, កែតម្រូវការរំខានថាមពលនៅក្នុងសាច់ដុំ, ធ្វើអោយប្រសើរឡើងនូវឈាមរត់គ្រឿងកុំព្យូទ័រ។ ថ្នាំដែលជំរុញមុខងារនៃប្រព័ន្ធសរសៃប្រសាទកណ្តាលក៏ត្រូវបានគេប្រើផងដែរ - cerebrolysin, nootropics ។ វិធីសាស្រ្តព្យាបាលដោយចលនាត្រូវបានគេប្រើយ៉ាងទូលំទូលាយជាពិសេស amplipulse - benzohexonium និង proserine phoresis ។ ក្នុងករណីខ្លះការកែតម្រូវ orthopedic នៃ contractures និង paresis គឺចាំបាច់។
លក្ខណៈវិនិច្ឆ័យនៃការពិនិត្យសុខភាព និងសង្គមរបស់ VUT
ការចង្អុលបង្ហាញសម្រាប់ VL ចំពោះអ្នកជំងឺដែលកំពុងធ្វើការ (ជាមួយ Kugelberg-Welander amyotrophy, amyotrophy ឆ្អឹងខ្នងចំពោះមនុស្សពេញវ័យ) អាចជាការដកសំណងបណ្តោះអាសន្នដែលជាវគ្គបង្ការនៃការព្យាបាលនៅក្នុងមន្ទីរពេទ្យ (រយៈពេល - 1-2 ខែ) ។
លក្ខណៈពិសេសនៃពិការភាព
ជាមួយនឹងទម្រង់កុមារភាពដែលរីកចម្រើនយ៉ាងឆាប់រហ័ស (ជំងឺ Werdnig-Hoffman) សមត្ថភាពក្នុងការផ្លាស់ទី និងការថែទាំខ្លួនឯងត្រូវបានចុះខ្សោយយ៉ាងឆាប់រហ័ស។ នៅក្នុងទម្រង់ផ្សេងទៀត សមត្ថភាពក្នុងការផ្លាស់ទី និងរៀបចំ (ទម្រង់ដាច់នៃ amyotrophy ចំពោះមនុស្សពេញវ័យ) និងការប្រាស្រ័យទាក់ទងគ្នាដោយសារតែ dysarthria (bulbospinal amyotrophy Kennedy) ទទួលរង។ នៅដំណាក់កាលចុងក្រោយនៃទម្រង់ទាំងអស់ លក្ខណៈវិនិច្ឆ័យឈានមុខគេសម្រាប់ការវាយតម្លៃស្ថានភាពនៃសកម្មភាពសំខាន់នឹងជាសមត្ថភាពក្នុងការផ្លាស់ទី និងការថែទាំដោយខ្លួនឯង ដោយសារតែការរីករាលដាលនៃ paresis និង contractures ។
ប្រភេទ contraindicated និងលក្ខខណ្ឌការងារ
1) គ្រប់ប្រភេទនៃកម្លាំងពលកម្មរាងកាយខ្លាំង ធ្វើការជាមួយទីតាំងរាងកាយដោយបង្ខំដោយមានភាពតានតឹងយូរនៃក្រុមសាច់ដុំជាក់លាក់មួយក្នុងល្បឿនដែលបានកំណត់ (នៅលើបន្ទាត់ជួបប្រជុំគ្នានៅក្នុងកងពលតូច) វិជ្ជាជីវៈបើកបរ; 2) ការងារទាក់ទងនឹងការប៉ះពាល់នឹងសារធាតុពុល រំញ័រ វិទ្យុសកម្ម។ល។
ការចង្អុលបង្ហាញសម្រាប់ការបញ្ជូនទៅកាន់ BMSE
1. ក្រោមអាយុ 18 ឆ្នាំនៅពេលដែលកុមារប្រសិនបើមានសូចនាករវេជ្ជសាស្ត្រអាចត្រូវបានគេទទួលស្គាល់ថាជាពិការ។
2. ការចុះខ្សោយកម្រិតមធ្យម ឬធ្ងន់ធ្ងរនៃមុខងារម៉ូទ័រ ការកំណត់សមត្ថភាពក្នុងការផ្លាស់ទីដោយឯករាជ្យ ការថែទាំខ្លួនឯង ឬការងារ។
3. អ្នកជំងឺដែលកំពុងធ្វើការមិនអាចបន្តធ្វើការក្នុងជំនាញរបស់ពួកគេបានទេ ដោយសារដំណើរការរីកចម្រើននៃជំងឺ ការឈប់សម្រាកបន្ត និងពិការភាពបណ្តោះអាសន្នរយៈពេលវែង។
អ្នកជំងឺដែលមានសមត្ថភាព
ជាមួយនឹងដំណើរវិវត្តន៍យឺត ៗ (ជាពិសេសជាមួយនឹងទម្រង់លេចធ្លោនៃមរតក); ជាមួយនឹង paresis ស្រាលនៃផ្នែកដាច់ស្រយាលនៃចុងទាបបំផុតឬតែផ្នែកខាងលើ (ឆ្អឹងខ្នង amyotrophy នៃមនុស្សពេញវ័យ); ជាមួយនឹង paresis ស្រាលនៅក្នុងចុងទាបជិតនិងក្រវាត់អាងត្រគាក, មានការអប់រំ, ជំនាញ, បទពិសោធន៍ការងារ, នៅក្នុងអវត្តមាននៃកត្តានៅក្នុងការងាររបស់ពួកគេដែលជះឥទ្ធិពលអវិជ្ជមានដល់ដំណើរនៃជំងឺនេះ។ នៅពេលបង្កើតលក្ខខណ្ឌការងារចាំបាច់សម្រាប់អ្នកជំងឺបែបនេះ ជារឿយៗយោងទៅតាមការសន្និដ្ឋានរបស់ក្រុមប្រឹក្សាត្រួតពិនិត្យស្ថាប័ន ពួកគេអាចបន្តធ្វើការបានយូរ។
ការពិនិត្យអប្បបរមាដែលត្រូវការនៅពេលយោងទៅ BMSE
1) ព័ត៌មានអំពីប្រភេទនៃមរតកនៃជំងឺ។
2) EMG, ENMG ។
3) លទ្ធផលនៃការពិនិត្យ pathomorphological នៃការធ្វើកោសល្យវិច័យសាច់ដុំ។
4) លទ្ធផលនៃការកំណត់សកម្មភាព CPK នៅក្នុងសេរ៉ូមឈាម។
ការចង្អុលបង្ហាញសម្រាប់ការបញ្ជូនទៅកាន់ BMSE
នៅពេលកំណត់អាយុក្រោម 18 ឆ្នាំ៖ ក) ការចុះខ្សោយផ្នែកនៃសកម្មភាពជីវិត និងការកែប្រែសង្គមមិនល្អដែលជាការបង្ហាញនៃជំងឺតំណពូជ (អាមីយ៉ូត្រូហ្វីតឆ្អឹងខ្នងរ៉ាំរ៉ៃនៃ Werdnig-Hoffmann, amyotrophy នៃ Kugelberg-Welander និងទម្រង់កុមារភាព និងអនីតិជនផ្សេងទៀត); ខ) ការចុះខ្សោយជាលំដាប់នៃមុខងារម៉ូទ័រនៃអវយវៈ។
នៅពេលកំណត់បន្ទាប់ពីអាយុ 18 ឆ្នាំ:
ក្រុម I: បានកំណត់នៅដំណាក់កាលក្រោយៗ ជាមួយនឹងការធ្វើឱ្យទូលំទូលាយពេញលេញនៃដំណើរការ atrophic: paraplegia ទាបឬខាងលើ, tetraparesis ធ្ងន់ធ្ងរ; ការខូចខាតយ៉ាងធ្ងន់ធ្ងរដល់សាច់ដុំនៃប្រម៉ោយ ការកន្ត្រាក់រីករាលដាលនៅក្នុងសន្លាក់ធំនៃអវយវៈចាប់តាំងពីជំងឺទាំងនេះនាំឱ្យមានការថយចុះនៃសមត្ថភាពក្នុងការផ្លាស់ទីនិងការថែទាំខ្លួនឯងនៃសញ្ញាបត្រទីបី (ជាធម្មតានៅក្នុងទម្រង់ Werdnig-Hoffman);
ក្រុមទី II: ការវិវត្តយឺតប៉ុន្តែនៅក្នុងដំណាក់កាលនៃការចាប់ផ្តើមទូទៅនៃដំណើរការ atrophic នៅក្នុងវត្តមាននៃ paraparesis ផ្នែកខាងក្រោមដែលបញ្ចេញសម្លេង ការរួមបញ្ចូលគ្នារបស់វាជាមួយ paraparesis កម្រិតមធ្យមនៃចុងខាងលើជាពិសេសប្រសិនបើដំណើរការនេះទាក់ទងនឹងកំភួនដៃ។ នៅក្នុងវត្តមាននៃ paraparesis ទាប proximal និង paresis នៃសាច់ដុំ girdle អាងត្រគាកដែលនាំឱ្យមានការថយចុះយ៉ាងខ្លាំងនៃសមត្ថភាពក្នុងការដើរនិងឈរ; ជាមួយ paraparesis ខាងលើបញ្ចេញសម្លេង - ទាំង distal និង proximal; នៅក្នុងករណីនៃការបន្ថែមសូម្បីតែជំងឺ bulbar កម្រិតស្រាលទៅនឹងការបញ្ចេញសំឡេងយ៉ាងខ្លាំង (2-3 ពិន្ទុ) paresis នៃជើង (យោងទៅតាមលក្ខណៈវិនិច្ឆ័យនៃការចុះខ្សោយនៃសមត្ថភាពក្នុងការផ្លាស់ទីសកម្មភាពការងារនៃសញ្ញាបត្រទីពីរ);
ក្រុមទី III៖ អាមីតូត្រូហ្វីសកំពុងរីកចម្រើនបន្តិចម្តងៗ ជាមួយនឹងប៉ារ៉ាសេសធ្ងន់ធ្ងរកម្រិតមធ្យមនៃចុងចុងខាងលើ ឬខាងក្រោម ភាពស្លេកស្លាំងធ្ងន់ធ្ងរកម្រិតមធ្យមនៃអវយវៈទាប និងក្រវាត់អាងត្រគាក ប្រសិនបើភាពខុសប្រក្រតីទាំងនេះនាំឱ្យអសមត្ថភាពក្នុងការធ្វើការក្នុងវិជ្ជាជីវៈចម្បង ហើយការងារសមហេតុផលគឺមិនអាចទៅរួចទេ ឬ សម្រាប់រយៈពេលនៃការបណ្តុះបណ្តាលវិជ្ជាជីវៈរបស់មនុស្សដែលគ្មានវិជ្ជាជីវៈក៏ដូចជាអ្នកដែលត្រូវបានបង្ខំឱ្យទទួលបានថ្មីមួយ (ជាធម្មតានៅក្នុងទម្រង់ Kugelberg-Welander) - យោងតាមលក្ខណៈវិនិច្ឆ័យនៃសមត្ថភាពមានកម្រិតចំពោះចលនាឯករាជ្យនៃសញ្ញាបត្រទី 1 និង (ឬ) ទៅ ការងារនៃសញ្ញាបត្រដំបូង។
ក្នុងករណីមានការចុះខ្សោយធ្ងន់ធ្ងរនៃមុខងារម៉ូទ័រ និងភាពឥតប្រយោជន៍នៃវិធានការស្តារនីតិសម្បទា ពិការភាពត្រូវបានបង្កើតឡើងដោយគ្មានកំណត់ (បន្ទាប់ពីការសង្កេតរបស់អ្នកជំងឺមិនលើសពី 4 ឆ្នាំ) ។
មូលហេតុនៃពិការភាព៖ 1) ពិការភាពតាំងពីកុមារភាព (ក្នុងទម្រង់ Kugelberg-Welander); 2) ជំងឺទូទៅ ឬជំងឺដែលទទួលបានអំឡុងពេលបម្រើយោធា (ជាមួយនឹងជំងឺឆ្អឹងខ្នងចំពោះមនុស្សពេញវ័យ)។
ត្រូវគ្នាទៅនឹងអ្វីដែលត្រូវបានអនុវត្តទាក់ទងនឹងអ្នកជំងឺដែលមាន PMD ។ សម្រាប់ទម្រង់ដែលមានទីតាំងហ្សែនដែលគេស្គាល់ ការធ្វើរោគវិនិច្ឆ័យមុនពេលសម្រាលគឺអាចធ្វើទៅបានក្នុងដំណាក់កាលដំបូងនៃការមានផ្ទៃពោះ។ នៅក្នុងករណីនៃ Kugelberg-Welander amyotrophy, ការការពារពិការភាពជាញឹកញាប់ត្រូវបានសម្របសម្រួលដោយការណែនាំអំពីអាជីពដំបូងនិងការអប់រំរបស់អ្នកជំងឺនៅក្នុង amyotrophy ឆ្អឹងខ្នងនៃមនុស្សពេញវ័យ, ការងារសមហេតុផល, ពេលខ្លះបន្ទាប់ពីទទួលបានវិជ្ជាជីវៈថ្មីមួយ។
ការស្តារនីតិសម្បទា
កម្មវិធីបុគ្គលត្រូវបានបង្កើតឡើងសម្រាប់អ្នកជំងឺដែលមានជំងឺ Kugelberg-Welander និង amyotrophies ឆ្អឹងខ្នងមនុស្សពេញវ័យ។
1. ការស្តារនីតិសម្បទាផ្នែកវេជ្ជសាស្រ្តមានវគ្គម្តងហើយម្តងទៀតនៃការព្យាបាលដោយថ្នាំ ការម៉ាស្សា ការព្យាបាលដោយរាងកាយ ការព្យាបាលនៅ sanatorium ការផ្គត់ផ្គង់ស្បែកជើង orthopedic ឧបករណ៍ជួសជុល គ្រែ និងពូកនៃការរចនាពិសេស។ ទិដ្ឋភាពផ្លូវចិត្តរបស់វារួមមានការរៀបចំការអប់រំសម្រាប់កុមារពិការនៅផ្ទះ ការណែនាំអ្នកជំងឺ និងឪពុកម្តាយរបស់គាត់ឆ្ពោះទៅរកការងារគ្រប់គ្រាន់ និងការរៀបចំជីវិតរបស់កុមារនៅក្នុងគ្រួសារ។
2. ការស្តារនីតិសម្បទា។
ក) ការបណ្តុះបណ្តាលវិជ្ជាជីវៈនៅក្នុងសាលាបច្ចេកទេស (lyceums, មហាវិទ្យាល័យ) មជ្ឈមណ្ឌលស្តារនីតិសម្បទាវិជ្ជាជីវៈសម្រាប់អ្នកជម្ងឺ Kugelberg-Welander amyotrophy និង amyotrophy ឆ្អឹងខ្នងចំពោះមនុស្សពេញវ័យ។ ជំនាញដែលបានណែនាំ៖ គណនេយ្យ ការគ្រប់គ្រងការិយាល័យ ទីផ្សារ នីតិសាស្រ្ត និងគណនេយ្យក្នុងប្រព័ន្ធសន្តិសុខសង្គម អ្នកបច្ចេកទេសស្តង់ដារ មេកានិកវិទ្យុសម្រាប់ជួសជុលឧបករណ៍វិទ្យុ និងទូរទស្សន៍ ជាងជួសជុលស្បែកជើង ជាងជួសជុលស្បែកជើង ជាងមើល ជាងចងសៀវភៅ។ល។
ខ) ការងាររបស់ជនពិការនៃក្រុម III ។ អាស្រ័យលើការធ្វើមូលដ្ឋានីយកម្មដ៏លេចធ្លោ និងភាពធ្ងន់ធ្ងរនៃសាច់ដុំ atrophy ការងារខាងក្រោមអាចត្រូវបានណែនាំ៖
- ពលកម្មផ្លូវចិត្ត៖ សេដ្ឋវិទូ អ្នករៀបចំផែនការ វិស្វករ អ្នកបច្ចេកទេសដំណើរការ មេធាវី អ្នកបកប្រែ ស្ថិតិ អ្នកសរសេរគន្ថនិទ្ទេស បណ្ណារក្ស។
- ការងារគណនេយ្យ ស្មៀន និងរដ្ឋបាល៖ គណនេយ្យករ អ្នកកំណត់ស្តង់ដារ គណនេយ្យករ អ្នកជំនួញ អធិការនាយកដ្ឋានធនធានមនុស្ស មេបញ្ជាការអន្តេវាសិកដ្ឋាន។ល។
- កម្លាំងពលកម្មរាងកាយស្រាល និងមធ្យម (សម្រាប់អ្នកជំងឺពិការជើង)៖ អ្នកដំឡើងផលិតផលខ្នាតតូច ជាងជួសជុលឧបករណ៍វិទ្យុ និងទូរទស្សន៍ក្នុងសិក្ខាសាលា អ្នកចងសៀវភៅ។
- ធ្វើការក្នុងវិជ្ជាជីវៈគ្រួសារធំ (ដោយការខូចខាតដល់ចុងខាងលើ ឬខាងក្រោម)៖ អ្នកយាម អ្នកចាំម៉ោង អ្នកបញ្ជូននៅកន្លែងធ្វើការនៅស្ថានី ប្រតិបត្តិករបញ្ជរ។
អនុសាសន៍ការងារគួរតែផ្តល់នូវលក្ខណៈល្អប្រសើរបំផុតនៃការងារដែលតម្រូវឱ្យមានភាពតានតឹងផ្នែករាងកាយនិងសរសៃប្រសាទស្រាលក្នុងស្ថានភាពសុខស្រួលឬជិតនឹងលក្ខខណ្ឌសុខស្រួល (ភាពធ្ងន់ធ្ងរប្រភេទ I - ការលើកទម្ងន់ម្តងមិនលើសពី 2 kcal សកម្មភាពរាងកាយក្នុងមួយវេន - 900 kcal) ។
គ) ជនពិការនៃក្រុម II (amyotrophy នៃមនុស្សពេញវ័យ និងជំងឺ Kugelberg-Welander) អាចត្រូវបានជួលនៅផ្ទះក្នុងជំនាញចម្បងរបស់ពួកគេ (បុគ្គលិកផ្លូវចិត្តដែលមានសមត្ថភាពខ្ពស់) ក៏ដូចជា bookbinder អ្នកស្អិត អ្នកប្រមូលផ្តុំផ្នែកតូចៗ ជាងដេរ។
3. ការស្តារនីតិសម្បទាសង្គម៖ ផ្តល់ជូនអ្នកជំងឺនូវឧបករណ៍ពិសេសសម្រាប់ប្រើប្រាស់បន្ទប់ទឹក បង្គន់ និងរទេះរុញ។ ដោយសារតែដំណើរវិវត្តន៍នៃជំងឺនេះ ការបើកបរត្រូវបាន contraindicated សម្រាប់អ្នកជំងឺ។
ជម្ងឺហ្សែនគឺជាជំងឺ pathological ដែលកើតឡើងដោយសារការផ្លាស់ប្តូរហ្សែន។ ជម្ងឺប្រភេទនេះត្រូវបានទទួលមរតក ហើយភាគច្រើនធ្វើឱ្យខ្លួនឯងមានអារម្មណ៍នៅវ័យកុមារភាព។ យោងតាមស្ថិតិនៅក្នុងពិភពសម័យទំនើប កុមារជាង 5 លាននាក់ក្នុងមួយឆ្នាំកើតមកមានជម្ងឺហ្សែនដោយសារតែការវិវត្តន៍ដែលកុមារជាច្រើនមិនរស់នៅរហូតដល់អាយុ 5 ឆ្នាំ។ ដូច្នេះហើយ សព្វថ្ងៃនេះ វាត្រូវបានគេបង្កើតឡើងថាប្រហែល 40-50% នៃមរណភាពកុមារភាពដំបូងគឺដោយសារតែការផ្លាស់ប្តូរតំណពូជ ដែលរួមមាន Kugelberg-Welander amyotrophy ។
Kugelberg-Welander amyotrophyគឺជាជំងឺសរសៃប្រសាទតំណពូជដែលបំផ្លាញសរសៃប្រសាទម៉ូទ័រនៃខួរឆ្អឹងខ្នង។ រោគវិទ្យានេះត្រូវបានចាត់ថ្នាក់ជាប្រភេទ III នៃខួរឆ្អឹងខ្នងអនីតិជន។ សញ្ញាដំបូងនៃ Kugelberg-Welander amyotrophy ជាធម្មតាលេចឡើងនៅចន្លោះអាយុពី 2 ទៅ 15 ឆ្នាំ។
បើប្រៀបធៀបជាមួយ amyotrophy ឆ្អឹងខ្នងនៃប្រភេទទារកនិងកម្រិតមធ្យម ជំងឺនេះមានការព្យាករណ៍អំណោយផលជាង - អាយុសង្ឃឹមរស់របស់អ្នកជំងឺដែលទទួលរងពី Kugelberg-Welander amyotrophy ត្រូវគ្នាទៅនឹងអត្រាមរណៈជាមធ្យមក្នុងចំណោមមនុស្សដែលមានសុខភាពល្អ។
ហេតុផល
ការអភិវឌ្ឍន៍នៃ Kugelberg-Welander amyotrophy ត្រូវបានធ្វើឱ្យសកម្មដោយសារតែការផ្លាស់ប្តូរនៅក្នុងអ្វីដែលគេហៅថា motor neuron survival gene (SMN1) ដែលមានទីតាំងនៅលើក្រូម៉ូសូម 5 និងទទួលខុសត្រូវចំពោះការផលិតប្រូតេអ៊ីនពិសេស កង្វះខាតដែលនាំទៅដល់ការស្លាប់របស់ម៉ូទ័រ។ កោសិកាសរសៃប្រសាទ។
មកទល់នឹងពេលនេះ វាត្រូវបានគេបង្ហាញថាមនុស្សប្រហែល 1 នាក់ក្នុងចំណោម 40 នាក់គឺជាអ្នកដឹកជញ្ជូននៃការផ្លាស់ប្តូរ DNA នេះ។ ដោយសារតែការពិតដែលថា amyotrophy ឆ្អឹងខ្នងគឺជារោគសាស្ត្រដែលមានប្រភេទ autosomal recessive នៃមរតក ការធ្វើឱ្យសកម្មរបស់វាអាចធ្វើទៅបានលុះត្រាតែឪពុកម្តាយទាំងពីរមានហ្សែនផ្លាស់ប្តូរដូចគ្នា។ ដូច្នេះប្រូបាប៊ីលីតេនៃការវិវត្តន៍ amyotrophy Kugelberg-Welander ចំពោះកុមារគឺមិនលើសពី 25% ទេ។
ជាក់លាក់
តាមក្បួនមួយរោគសញ្ញាដំបូងនៃ Kugelberg-Welander amyotrophy ចាប់ផ្តើមលេចឡើងនៅអាយុ 2 ឆ្នាំហើយនៅអាយុក្រោយជំងឺនេះធ្វើឱ្យខ្លួនឯងមានអារម្មណ៍កម្រណាស់។
ដោយសារតែភាពទន់ខ្សោយនៃសាច់ដុំនៅក្នុងប្រភេទ III ឆ្អឹងខ្នង amyotrophy មានការរីកចម្រើនបន្តិចម្តងៗ សមត្ថភាពសម្រាប់ចលនាឯករាជ្យ និងការថែទាំដោយខ្លួនឯងចំពោះអ្នកជំងឺដែលទទួលរងពីរោគសាស្ត្រនេះត្រូវបានរក្សាទុកក្នុងរយៈពេលយូរ។
តាមក្បួនមួយ មនុស្សដែលត្រូវបានគេធ្វើរោគវិនិច្ឆ័យថាមាន Kugelberg-Welander amyotrophy ត្រូវបានចាត់តាំងជាក្រុមពិការភាព ដែលកម្រិតត្រូវបានកំណត់ដោយយោងទៅតាមភាពធ្ងន់ធ្ងរនៃការចុះខ្សោយនៃម៉ូទ័រ ពិការភាព និងការពឹងផ្អែកលើមនុស្សផ្សេងទៀត។
វាជាការលំបាកខ្លាំងណាស់ក្នុងការទស្សន៍ទាយថាតើជំងឺនេះនឹងប៉ះពាល់ដល់ស្ថានភាពរបស់កុមារយ៉ាងដូចម្តេច។ ដូច្នេះហើយ អ្នកជំងឺខ្លះក្លាយទៅជាពិការយ៉ាងខ្លាំងសូម្បីតែក្នុងវ័យជំទង់ ហើយខ្លះអាចរក្សាបាននូវសមត្ថភាពសម្របសម្រួលដែលត្រូវបានអភិវឌ្ឍយ៉ាងល្អ សូម្បីតែនៅអាយុ 40 ឆ្នាំ។
រោគសញ្ញា
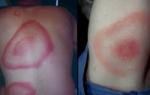
Kugelberg-Welander amyotrophy បង្ហាញខ្លួនជាចម្បងតាមរយៈការបង្កើនភាពទន់ខ្សោយនៃសាច់ដុំនៅជើង ដែលអាចធ្វើឱ្យកុមារមានការដើរហើរ។ លើសពីនេះទៀត កុមារដែលទទួលរងពីជំងឺឆ្អឹងខ្នង សូម្បីតែបន្ទាប់ពីដើរខ្លី និងរត់ចម្ងាយខ្លីក៏ដោយ ក៏ត្អូញត្អែរពីភាពអស់កម្លាំង។ កុមារដែលមានជំងឺនេះច្រើនតែដួល បាត់បង់លំនឹង និងពិបាកឡើងជណ្តើរ។ លេចឡើងដំបូងនៅក្នុងសាច់ដុំនៃជើងនិងឆ្អឹងអាងត្រគាក, អារម្មណ៍នៃភាពទន់ខ្សោយបន្តិចម្តងផ្លាស់ទីទៅជាលិកាសាច់ដុំនៃតំបន់ស្មានិងកែងដៃ។
នៅពេលដែលជំងឺនេះរីកចម្រើន ការធ្លាក់ទឹកចិត្តម៉ូតូនឹងវិវឌ្ឍន៍នៅក្នុងសាច់ដុំមុខ។ លើសពីនេះទៀត ការឆ្លុះសរសៃពួរបានបាត់ ហើយការកន្ត្រាក់ដោយអចេតនានៃសាច់ដុំដៃកើតឡើង។
រោគសញ្ញាដែលបង្ហាញពីការធ្វើឱ្យសកម្មនៃ Kugelberg-Welander amyotrophy ក៏រួមបញ្ចូលការកន្ត្រាក់អណ្តាត និងការថយចុះនៃចលនានៃសន្លាក់ស្មា និងកែងដៃ។ ជាលទ្ធផលនៃការ atrophy នៃសាច់ដុំនៃ girdle ស្មា, អ្នកជំងឺវិវត្តទៅជារោគសញ្ញានៃ blade ស្លាប។
ក្នុងករណីជាច្រើន កុមារដែលទទួលរងពីជំងឺនេះ ជួបប្រទះនឹងភាពមិនពិតនៃសាច់ដុំកំភួនជើង និងគូទ ដែលត្រូវបានបង្កើតឡើងដោយការជំនួសជាលិកាសាច់ដុំជាមួយនឹងជាលិកាខ្លាញ់ និងជាលិកាភ្ជាប់។
នៅពេលដែល Kugelberg-Welander amyotrophy រីកចម្រើន ការខូចទ្រង់ទ្រាយនៃរចនាសម្ព័ន្ធឆ្អឹងនៃប្រព័ន្ធ musculoskeletal ក៏អាចកើតមានផងដែរ រួមទាំងការកោងនៃឆ្អឹងខ្នង ការខូចទ្រង់ទ្រាយនៃទ្រូង និងជើង។
នៅដំណាក់កាលក្រោយនៃការអភិវឌ្ឍន៍នៃ Kugelberg-Welander amyotrophy ការបរាជ័យផ្លូវដង្ហើមជាធម្មតាវិវឌ្ឍន៍ដែលបណ្តាលឱ្យហើមផ្លូវដង្ហើមនិងដង្ហើមខ្លីដែលប្រែទៅជាថប់ដង្ហើមបន្តិចម្តង ៗ ។ កង្វះការថែទាំវេជ្ជសាស្រ្តសង្គ្រោះបន្ទាន់សម្រាប់ស្ថានភាពរោគសាស្ត្របែបនេះគឺពោរពេញទៅដោយការស្លាប់។
រោគវិនិច្ឆ័យ
វាអាចធ្វើទៅបានដើម្បីបញ្ជាក់ការធ្វើរោគវិនិច្ឆ័យក្នុងករណីមានការសង្ស័យថាមានការធ្វើឱ្យសកម្មនៃ Kugelberg-Welander amyotrophy តាមរយៈការធ្វើតេស្ត DNA ។ Electroneuromyography ក៏ត្រូវបានចេញវេជ្ជបញ្ជាជានីតិវិធីវិនិច្ឆ័យបន្ថែម ដោយមានជំនួយពីភាពតានតឹងសាច់ដុំត្រូវបានត្រួតពិនិត្យកម្រិតនៃសកម្មភាពនៃណឺរ៉ូនម៉ូទ័រត្រូវបានវាយតម្លៃ ហើយវត្តមាននៃការផ្លាស់ប្តូរ denervation ត្រូវបានកំណត់។ លើសពីនេះទៀត ប្រសិនបើចាំបាច់ ការធ្វើកោសល្យវិច័យសាច់ដុំគ្រោងឆ្អឹងអាចត្រូវបានអនុវត្ត។
ការព្យាបាល
សម្រាប់អ្នកជំងឺដែលទទួលរងពី Kugelberg-Welander amyotrophy ការព្យាបាលតាមរោគសញ្ញាត្រូវបានចង្អុលបង្ហាញ ដែលក្នុងនោះវគ្គព្យាបាលពិសេសត្រូវបានបង្កើតឡើងក្នុងគោលបំណងកាត់បន្ថយការបង្ហាញរោគសញ្ញានៃជំងឺនេះ។ ជាដំបូងអ្នកជំងឺដែលមានរោគវិនិច្ឆ័យនេះត្រូវបានចេញវេជ្ជបញ្ជាថ្នាំ nootropic, anabolic steroids និងវីតាមីនស្មុគស្មាញ។
វិធីសាស្រ្តដ៏មានប្រសិទ្ធភាពក្នុងការបន្ធូរបន្ថយស្ថានភាពអ្នកជំងឺនៃទម្រង់នេះក៏រួមបញ្ចូលការធ្វើលំហាត់ប្រាណព្យាបាល និងការម៉ាស្សាស្តារឡើងវិញផងដែរ។ លើសពីនេះទៀត ការកែតម្រូវឆ្អឹងអាចត្រូវបានណែនាំសម្រាប់អ្នកជំងឺដែលមានការខូចទ្រង់ទ្រាយឆ្អឹងធ្ងន់ធ្ងរ។
អាមីតូត្រូហ្វីតឆ្អឹងខ្នង Kugelberg-Welander
ឈ្មោះមួយផ្សេងទៀតសម្រាប់ជំងឺនេះគឺ amyotrophy ឆ្អឹងខ្នងស្លូតបូតនៃកុមារភាពនិងវ័យជំទង់ជាមួយនឹងការ fasciculations ។ ប្រភេទនៃមរតកគឺ autosomal recessive ។ រោគសញ្ញាដំបូងនៃជំងឺនេះអាចលេចឡើងក្នុងវ័យកុមារភាព ប៉ុន្តែភាគច្រើនជាញឹកញាប់នៅអាយុ 8-12 ឆ្នាំ។ លក្ខណៈដោយការកន្ត្រាក់នៃសាច់ដុំនៃអវយវៈជិតៗ ការរមួលសាច់ដុំ និងសរសៃពួរ។ ភាពយឺតយ៉ាវនៃសាច់ដុំ និងភាពយឺតយ៉ាវក្នុងការអភិវឌ្ឍន៍រាងកាយ និងផ្លូវចិត្ត ត្រូវបានគេសង្កេតឃើញជាញឹកញាប់។
អាមីតូត្រូហ្វីតសរសៃប្រសាទនៃ Charcot-Marie-Tooth
វាត្រូវបានទទួលមរតកក្នុងលក្ខណៈ autosomal លេចធ្លោ ហើយចាប់ផ្តើមជាញឹកញាប់បំផុតនៅអាយុ 20-30 ឆ្នាំ មិនសូវជាញឹកញាប់នៅអាយុសិក្សា និងមានប្រភេទរង 1a និង 16។ ហ្សែននេះត្រូវបានគេផ្គូផ្គងទៅនឹងដៃវែងនៃក្រូម៉ូសូម ៥។ ការក្លូនតាមទីតាំងបានបង្ហាញពីទីតាំងជំងឺនៅលើក្រូម៉ូសូម 17p11.2 (ប្រភេទ 1a) និងទីតាំងតូចជាងនៅលើក្រូម៉ូសូម Ig22 (ប្រភេទ 16)។
Pathomorphologically, amyotrophy សរសៃប្រសាទត្រូវបានកំណត់លក្ខណៈដោយការផ្លាស់ប្តូរ degenerative នៅក្នុងស្នែងផ្នែកខាងមុខនិងជួរឈរក្រោយនៃខួរឆ្អឹងខ្នងក៏ដូចជានៅក្នុងឫសនិងសរសៃប្រសាទគ្រឿងកុំព្យូទ័រ។
ការបង្ហាញគ្លីនិក។ ការផ្លាស់ប្តូរ Atrophic នៅក្នុងសាច់ដុំនៃចុងចុងបំផុត ដែលភាគច្រើនបំផុតគឺផ្នែកខាងក្រោម គ្របដណ្តប់។ ផ្នែកបន្ថែមនៃជើងទាប ក៏ដូចជាការបត់ជើងធំនៃជើងត្រូវបានប៉ះពាល់ ដែលជាលទ្ធផលដែលជើងចាប់ផ្តើមស្រក។ អ្នកជំងឺដើរលើកជង្គង់របស់គាត់ឱ្យខ្ពស់ (ជំហាន) ហើយទីតាំង valgus នៃជើងមានការរីកចម្រើន (ការបង្វិលខាងក្រៅ) ។ ការឆ្លុះសរសៃពួរដែលភាគច្រើនជាញឹកញាប់ Achilles បាត់ទៅវិញ។ មានភាពខុសប្លែកគ្នាខ្លះរវាងការដាច់សាច់ដុំសំខាន់ៗ និងការរក្សាមុខងារម៉ូទ័រ ក្នុងករណីភាគច្រើន ភាពខុសប្រក្រតីនៃប្រភេទ "ស្រោមដៃ" និង "ស្រោមជើង" ត្រូវបានកត់សម្គាល់។ ការឈឺចាប់ និង paresthesia អាចនឹងលេចឡើង ក៏ដូចជាការថយចុះនៃភាពប្រែប្រួលខ្លាំង ដោយសារតែការខូចខាតដល់ជួរឈរក្រោយនៃខួរឆ្អឹងខ្នង។ ការខូចទ្រង់ទ្រាយនៃជើងត្រូវបានរកឃើញជាញឹកញាប់: ពួកវាក្លាយទៅជាប្រហោងជាមួយនឹងក្លោងខ្ពស់ផ្នែកបន្ថែមនៃមេនិងការបត់បែននៃ phalanges ស្ថានីយ។ ដំណើរការនៃជំងឺនេះមានការវិវឌ្ឍយឺតៗ។
ជំងឺនេះកើតឡើងក្នុងវ័យកុមារភាព ហើយត្រូវបានកំណត់លក្ខណៈដោយវគ្គសាហាវជាមួយនឹងការវិវត្តន៍យ៉ាងឆាប់រហ័ស។ អាស្រ័យលើពេលវេលានៃការលេចឡើងនៃរោគសញ្ញាដំបូងនិងអត្រានៃការរីកលូតលាស់នៃដំណើរការនេះទម្រង់បីនៃជំងឺត្រូវបានសម្គាល់: ពីកំណើត, កុមារភាពដំបូងនិងចុង។ទម្រង់ពីកំណើតអាចលេចឡើងក្នុងអំឡុងពេលមានផ្ទៃពោះ។ ក្នុងករណីបែបនេះ ចលនាគភ៌ដែលមានលក្ខណៈធម្មតានៅពេលដំបូង ចុះខ្សោយនៅដំណាក់កាលក្រោយនៃការមានផ្ទៃពោះ ការសម្រាលកូនអាចជារោគសាស្ត្រ ហើយរួចទៅហើយនៅក្នុងថ្ងៃដំបូងបន្ទាប់ពីកំណើតនៃកូន ភាពស្លេកស្លាំងនៃសាច់ដុំជាក់ស្តែងត្រូវបានរកឃើញជាមួយនឹងការថយចុះនៃសាច់ដុំ។ សម្លេង និងការថយចុះនៃការឆ្លុះសរសៃពួរ។ ពេលខ្លះ areflexia ពេញលេញត្រូវបានកត់សម្គាល់។ រោគសញ្ញានៃដុំពកដំបូងអាចកើតមានឡើង ដែលបង្ហាញដោយការយំខ្សោយ និងការជញ្ជក់យឺតៗ។ ចំពោះកុមារ, ជំងឺ fibrillation នៅក្នុងអណ្តាត, ការថយចុះនៃការឆ្លុះបញ្ចាំង pharyngeal និង hypomimia អាចត្រូវបានរកឃើញ។ Tachycardia ជាធម្មតាត្រូវបានគេសង្កេតឃើញ។ ជំងឺនេះច្រើនតែរួមផ្សំជាមួយនឹងពិការភាពក្នុងការអភិវឌ្ឍន៍ និងវិកលចរិតមួយចំនួន។ វគ្គនៃជំងឺនេះគឺលឿនណាស់ការស្លាប់កើតឡើងដោយ 1-l.5 ឆ្នាំ។
ទម្រង់កុមារភាពដំបូងលក្ខណៈដោយវគ្គសិក្សាស្រាលជាងបន្តិចបើប្រៀបធៀបទៅនឹងពីកំណើត។ ទម្រង់នេះត្រូវបានគេចាត់ទុកថាជាបុរាណ។ ការចាប់ផ្តើមនៃជំងឺនេះកើតឡើងមុនអាយុ 1,5 ឆ្នាំ។ ក្នុងករណីភាគច្រើន រោគសញ្ញាដំបូងលេចឡើងបន្ទាប់ពីការឆ្លងមួយចំនួន ឬការពុលអាហារ។ កុមារដែលពីមុនមានការអភិវឌ្ឍន៍ច្រើន ឬតិចជាងធម្មតា បាត់បង់ជំនាញម៉ូតូដែលបានទទួលពីមុនយ៉ាងឆាប់រហ័ស ហើយឈប់ដើរ ឈរ ឬអង្គុយ។ Flaccid paresis ដំបូងកើតឡើងនៅក្នុងជើង បន្ទាប់មកនៅក្នុងសាច់ដុំនៃដងខ្លួន និងដៃ។ ស្ថានភាពកាន់តែយ៉ាប់យ៉ឺនយ៉ាងឆាប់រហ័ស ភាពទន់ខ្សោយលេចឡើងនៅក្នុងសាច់ដុំក និងសាច់ដុំ bulbar ។ នៅអាយុ 4-5 ឆ្នាំជាធម្មតាជាលទ្ធផលនៃការបរាជ័យផ្លូវដង្ហើម, ជំងឺរលាកសួតកើតឡើងហើយការស្លាប់កើតឡើង។ ចំពោះអ្នកជំងឺ, paresis flaccid ត្រូវបានអមដោយការវិវត្តនៃការកន្ត្រាក់សរសៃពួរ។ hyperhidrosis ទូទៅត្រូវបានគេសង្កេតឃើញជាញឹកញាប់។
ទម្រង់យឺតចាប់ផ្តើមនៅអាយុ 1.5-2 ឆ្នាំហើយហូរបានយ៉ាងងាយស្រួលបើប្រៀបធៀបទៅនឹងទម្រង់ពីរដំបូង។ អ្នកជំងឺរហូតដល់ 10 ឆ្នាំអាចរក្សាសមត្ថភាពក្នុងការផ្លាស់ទី។
រោគសញ្ញានាំមុខគេគឺ paresis នៅផ្នែកជិតៗនៃជើង បន្ទាប់មកដៃ។ សាច់ដុំ atrophy គឺពិបាកក្នុងការរកឃើញដោយសារតែស្រទាប់ខ្លាញ់ subcutaneous ដែលបានកំណត់យ៉ាងល្អ។ ការឆ្លុះសរសៃពួរសរសៃពួរបាត់មុន។ លក្ខណៈគឺជាការញ័រម្រាមដៃនៃដៃដែលលាតចេញ (ញ័រ fascicular) ។ ការខូចទ្រង់ទ្រាយឆ្អឹងគឺជារឿងធម្មតា ជាពិសេសនៅក្នុងទ្រូង ប៉ុន្តែក៏មាននៅផ្នែកខាងក្រោមផងដែរ។ រោគសញ្ញា Bulbar ត្រូវបានតំណាងដោយការចុះខ្សោយនៃសាច់ដុំអណ្តាតជាមួយនឹងការកន្ត្រាក់ fibrillary, paresis នៃក្រអូមមាត់ទន់ជាមួយនឹងការថយចុះនៃការឆ្លុះបញ្ចាំង pharyngeal ។
វ៉ារ្យ៉ង់ពិសេសនៃជម្ងឺឆ្អឹងខ្នង Werdnig-Hoffmann ត្រូវបានគេស្គាល់ថា - ពិការភាពរីកចម្រើនឬជំងឺ Fazio-Londe ។ ជំងឺនេះច្រើនតែចាប់ផ្តើមនៅចុងបញ្ចប់នៃឆ្នាំទី 2 នៃជីវិត ជួនកាលក្នុងវ័យជាអនីតិជន ហើយត្រូវបានកំណត់លក្ខណៈដោយភាពទន់ខ្សោយនៃសាច់ដុំមុខ រួមទាំងសាច់ដុំ masticatory ពិបាកលេប ការផ្លាស់ប្តូរសំលេង និងការដាច់រលាត់នៃសាច់ដុំអណ្តាត។ . Ophthalmoplegia អាចកើតឡើង។ ជំងឺនេះរីកចម្រើនយ៉ាងឆាប់រហ័សហើយការស្លាប់កើតឡើង 6-12 ខែបន្ទាប់ពីការចាប់ផ្តើមនៃរោគសញ្ញាដំបូង។ ជម្ងឺ Bulbar អាចត្រូវបានអមដោយ paresis flaccid និងខ្វិននៃអវយវៈពេលខ្លះពួកគេមិនមានពេលវេលាដើម្បីអភិវឌ្ឍទេប៉ុន្តែនៅពេលធ្វើកោសល្យវិច័យការខូចខាតដល់កោសិកានៃស្នែងផ្នែកខាងមុខនៃខួរឆ្អឹងខ្នងត្រូវបានរកឃើញជានិច្ចនៅទូទាំងប្រវែងរបស់វា។ ករណីគ្រួសារនៃជំងឺ Fazio-Londe ត្រូវបានពិពណ៌នា នៅពេលដែលបងប្អូនបង្កើតម្នាក់ ឬច្រើននាក់បានរងទុក្ខ។ ប្រភេទនៃការបញ្ជូនបន្តពូជគឺ autosomal recessive ។
ការធ្វើរោគវិនិច្ឆ័យនៃ amyotrophy ឆ្អឹងខ្នងរបស់ Werdnig-Hoffmannត្រូវបានផ្អែកលើ (បន្ថែមពីលើការចាប់ផ្តើមដំបូងនៃជំងឺនេះនិងរូបភាពគ្លីនិកលក្ខណៈ) លើលទ្ធផលនៃវិធីសាស្រ្តស្រាវជ្រាវបន្ថែមដែលក្នុងនោះ electromyography គួរតែត្រូវបានចង្អុលបង្ហាញជាមុន។ សកម្មភាពជីវអគ្គិសនីដោយឯកឯងនៅពេលសម្រាកជាមួយនឹងវត្តមាននៃសក្តានុពល fasciculation ស្ទើរតែតែងតែត្រូវបានរកឃើញ។ ក្នុងអំឡុងពេលនៃការកន្ត្រាក់ដោយស្ម័គ្រចិត្ត សកម្មភាពអគ្គិសនីយឺតជាមួយនឹង "ចង្វាក់របងជ្រើសរើស" ត្រូវបានកត់ត្រា ដែលបង្ហាញពីបាតុភូតនៃការធ្វើសមកាលកម្ម និងការកើនឡើងនៃរយៈពេលនៃសក្តានុពល។
ការពិនិត្យរោគសាស្ត្របង្ហាញពីការថយចុះនៃចំនួនកោសិកានៅក្នុងស្នែងខាងមុខនៃខួរឆ្អឹងខ្នង និងការផ្លាស់ប្តូរ degenerative នៅក្នុងពួកវា។ ការផ្លាស់ប្តូររោគសាស្ត្រត្រូវបានប្រកាសជាពិសេសនៅក្នុងតំបន់នៃ lumbar និង cervical thickenings ក៏ដូចជានៅក្នុងស្នូលម៉ូទ័រនៃសរសៃប្រសាទ cranial ។ ការផ្លាស់ប្តូរត្រូវបានរកឃើញនៅក្នុងឫសខាងមុខ និងនៅក្នុងផ្នែក intramuscular នៃចុងសរសៃប្រសាទ។ ក្រោយមកទៀត ស្ថានីយធម្មតារលាយបាត់ ហើយក្លាយជាសាខាហួសប្រមាណ។
ការសិក្សាជីវគីមីបង្ហាញពីការផ្លាស់ប្តូរនៃការរំលាយអាហារកាបូអ៊ីដ្រាត។ ដូច្នេះ E. A. Savelyeva-Vasilieva (1973) បានរកឃើញថា glycolysis ចំពោះអ្នកជំងឺដែលមាន amyotrophy ឆ្អឹងខ្នង Werdnig-Hoffmann ខិតជិតប្រភេទអំប្រ៊ីយ៉ុង។ ជាញឹកញាប់ការរំខានដ៏សំខាន់ក្នុងការរំលាយអាហារ creatine-creatinine ត្រូវបានរកឃើញ - ការកើនឡើងនៃការបញ្ចេញ creatine ក្នុងទឹកនោមការថយចុះការបញ្ចេញ creatinine ។ វាជាការសំខាន់ក្នុងការកត់សម្គាល់ថាកម្រិតនៃអង់ស៊ីមនៅក្នុងសេរ៉ូមឈាមនៅតែស្ទើរតែមិនផ្លាស់ប្តូរ។
អាមីតូត្រូហ្វីតឆ្អឹងខ្នង Werdnig-Hoffmannសំដៅទៅលើជំងឺតំណពូជជាមួយនឹងប្រភេទ autosomal recessive នៃការបញ្ជូន។ ពិការភាពជីវគីមីចម្បងគឺមិនស្គាល់។ មានការសន្មត់ថាពិការភាពហ្សែននាំទៅរកការបង្កើតកោសិកានៃស្នែងមុននៃខួរឆ្អឹងខ្នងការរំខាននៃភាពខុសគ្នារបស់ពួកគេនិងការអភិវឌ្ឍតិចតួចនៃអ្នកទទួល cholinergic សាច់ដុំ។
នៅពេលធ្វើរោគវិនិច្ឆ័យ Werdnig-Hoffmann amyotrophy ឆ្អឹងខ្នង ភាពខុសគ្នាត្រូវបានធ្វើឡើងជាមួយ Oppenheim myotonia ។ យោងតាមអ្នកស្រាវជ្រាវភាគច្រើន Oppenheim myotopia មិនមែនជាអង្គភាព nosological ឯករាជ្យនោះទេប៉ុន្តែជារោគសញ្ញាដែលជាការបង្ហាញនាំមុខគេដែលត្រូវបានប្រកាសថា hypotonia សាច់ដុំ។ ក្នុងន័យនេះ ពាក្យថា "ទារកទន់ខ្សោយ" ឬ "កូនទន់ខ្សោយ" ឥឡូវនេះបានរីករាលដាល។ រោគសញ្ញា "កូនមិនស្អាត" ត្រូវបានគេសង្កេតឃើញនៅក្នុងជំងឺដូចជាជំងឺសាច់ដុំពីកំណើត ទម្រង់ស្លូតបូតនៃ hypotonia ពីកំណើត rickets ទម្រង់អាតូនិចនៃជំងឺខួរក្បាល ក៏ដូចជាការរងរបួសឆ្អឹងខ្នងឆ្លង ជម្ងឺខួរឆ្អឹងខ្នងស្រួចស្រាវក្នុងស្បូន ឬ polyradiculoneuritis ។ រោគសញ្ញា "កូនមិនស្អាត" អាចកើតមានឡើងជាមួយនឹងជំងឺសាច់ដុំសកល (ជំងឺ Krabbe) ជាមួយនឹងជំងឺ glycogenosis ជាពិសេសជាមួយនឹងប្រភេទ II ឬជំងឺ Pompe (glycogenosis សកល) ។
ការព្យាបាលសម្រាប់អាមីយ៉ូត្រូហ្វីតឆ្អឹងខ្នង Werdnig-Hoffmann វាមកលើការចេញវេជ្ជបញ្ជាម៉ាស្សា និងការព្យាបាលដោយលំហាត់ប្រាណ ដែលគួរតែត្រូវបានអនុវត្តជាប្រព័ន្ធ។ មិនមានការព្យាបាលរ៉ាឌីកាល់ទេ។
ភាពប្រសើរឡើងមួយចំនួនត្រូវបានផ្តល់ដោយថ្នាំដូចជា Cerebrolysin, aminalon, ថ្នាំ anticholinesterase (proserin, oxazil, galantamine, sanguinarine), វីតាមីន B ការចាក់ម្តងហើយម្តងទៀតនៃឈាមក្រុមដូចគ្នា (50 មីលីលីត្រ 4-5 ដង) ត្រូវបានគេចាត់ទុកថាជាទូទៅ។ ភ្នាក់ងារពង្រឹង និងត្រូវបានចង្អុលបង្ហាញនៅក្នុងដំណាក់កាលច្បាស់លាស់នៃជំងឺ។
ទម្រង់ Pseudomyopathic នៃ amyotrophy ឆ្អឹងខ្នងរីកចម្រើន Kugelberg-Welander
នៅឆ្នាំ 1942 Wohlfart ដំបូងបានពិពណ៌នាអំពីជំងឺដែលបង្ហាញដោយសាច់ដុំ atrophies និង paresis និងស្រដៀងទៅនឹងជំងឺសាច់ដុំបឋម ប៉ុន្តែជាមួយនឹងការរីករាលដាលយ៉ាងទូលំទូលាយ។ នៅឆ្នាំ 1956 Kugelberg និង Welander បានសង្កត់ធ្ងន់ថាជំងឺបែបនេះគឺមានលក្ខណៈស្លូតបូត។ ការត្រួតពិនិត្យ electromyographic ដោយប្រុងប្រយ័ត្នបានអនុញ្ញាតឱ្យអ្នកនិពន្ធបញ្ជាក់ពីធម្មជាតិ neurogenic នៃសាច់ដុំ atrophy និងចាត់ថ្នាក់ចុងក្រោយថាជាដំបៅឆ្អឹងខ្នង។ជំងឺនេះចាប់ផ្តើមក្នុងករណីភាគច្រើននៅអាយុ 3-6 ឆ្នាំ ហើយវិវត្តន៍យឺតណាស់។ ករណីនៃការលេចឡើងក្រោយនៃរោគសញ្ញាដំបូង រួមទាំងចំពោះមនុស្សពេញវ័យក៏ត្រូវបានពិពណ៌នាផងដែរ។ អ្នកជំងឺរក្សាសមត្ថភាពក្នុងការថែទាំខ្លួនឯងបានយូរ និងសូម្បីតែពេលខ្លះធ្វើការ។ យោងតាមរោគសញ្ញាគ្លីនិក ជំងឺនេះប្រហាក់ប្រហែលនឹងទម្រង់អវយវៈ-ក្រវាត់ (ជំងឺសាច់ដុំរបស់ Erb)។ ភាពទន់ខ្សោយនៃសាច់ដុំ និងដុំពកកើតឡើងដំបូងនៅផ្នែកជិតៗនៃចុងទាបបំផុត និងក្រវាត់អាងត្រគាក បន្ទាប់មករីករាលដាលដល់ខ្សែស្មា។ ភាពស្រដៀងគ្នាជាមួយនឹងជំងឺសាច់ដុំរបស់ Erb ត្រូវបានបំពេញបន្ថែមដោយវត្តមាននៃ pseudohypertrophy នៃសាច់ដុំ gastrocnemius នៅក្នុងករណីមួយចំនួនធំ។ ការខូចទ្រង់ទ្រាយឆ្អឹង និងការដកសរសៃពួរចេញជាធម្មតាគឺអវត្តមាន។ ជាមួយនឹង Kugelberg-Welander amyotrophy ដំណើរការអាចរីករាលដាលទៅតំបន់ bulbar ដែលត្រូវបានបង្ហាញតាមគ្លីនិកដោយការថយចុះនៃអណ្តាត និងការរមួលសាច់ដុំ។ ក្រោយមកទៀតក៏អាចត្រូវបានគេសង្កេតឃើញនៅក្នុងសាច់ដុំមុខ។ ជំងឺម៉ូទ័រដែលជាការបង្ហាញនៃការខូចខាតនុយក្លេអ៊ែរចំពោះគូ X-IX-XII និង VII នៃសរសៃប្រសាទ cranial ត្រូវបានរកឃើញយឺតណាស់តែនៅដំណាក់កាលជឿនលឿននៃដំណើរការរោគសាស្ត្រ។
ការសិក្សាបន្ថែមនៅក្នុង Kugelberg-Welander spinal amyotrophy បង្ហាញពីការផ្លាស់ប្តូរដ៏ចម្លែក - electromyography បង្ហាញពីសញ្ញាច្បាស់លាស់នៃការខូចខាតឆ្អឹងខ្នង ក្នុងពេលជាមួយគ្នានោះ រូបភាព pathomorphological អំឡុងពេលធ្វើកោសល្យវិច័យសាច់ដុំត្រូវបានតំណាងដោយលក្ខណៈចម្រុះនៃរោគសាស្ត្រ - រួមជាមួយនឹង neurogenic amyotrophy មានសូចនាករ នៃសញ្ញា dystrophic មួយចំនួន។ ទិន្នន័យស្រដៀងគ្នានេះត្រូវបានទទួលបានពីការសិក្សាជីវគីមី - សកម្មភាពនៃអង់ស៊ីមរួមទាំង creatine phosphokinase ត្រូវបានកើនឡើងជាញឹកញាប់ទោះបីជាក្នុងកម្រិតតិចជាងជាមួយនឹងជំងឺ myopathy ពិតក៏ដោយ។ សូចនាករនៃការផ្លាស់ប្តូរការរំលាយអាហារ creatine-creatinine ។
Kugelberg-Welander spinal amyotrophy គឺជាជំងឺតំណពូជដែលមានប្រភេទ autosomal recessive នៃការបញ្ជូន ហើយតាមមើលទៅ ជាមួយនឹងការជ្រៀតចូលមិនពេញលេញ ចាប់តាំងពីករណីកើតឡើងជាញឹកញាប់។ មានការពិពណ៌នាខ្លះអំពីមរតក autosomal លេចធ្លោនៃជំងឺនេះ។ រហូតមកដល់ពេលនេះ មិនមែនអ្នកនិពន្ធទាំងអស់ចាត់ទុក Kugelberg-Welandeoa amyotrophy ជាជំងឺឯករាជ្យទេ ដោយចាត់ទុកវាគ្រាន់តែជាការប្រែប្រួល "ស្រាល" នៃជំងឺ Werdnig-Hoffmann ប៉ុណ្ណោះ។ អាគុយម៉ង់ចម្បងក្នុងការពេញចិត្តនៃសេចក្តីថ្លែងការណ៍នេះគឺការសង្កេតនៅក្នុងគ្រួសារមួយនៃបងប្អូនបង្កើតដែលមានទម្រង់ទាំងពីរនៃ amyotrophy ឆ្អឹងខ្នង។ ទោះបីជាយ៉ាងណាក៏ដោយ វត្តមាននៃរោគសញ្ញាដូចជាសាច់ដុំ pseudohypertrophy, hyperfermentemia និងវគ្គសិក្សាស្រាលជាពិសេសផ្តល់សក្ខីកម្មដល់ឯករាជ្យភាព nosological នៃ Kugelberg-Welander amyotrophy ។ តាមទស្សនៈជាក់ស្តែង នេះមានសារៈសំខាន់ ដោយសារមានការព្យាករណ៍ខុសគ្នាសម្រាប់ទម្រង់ទាំងពីរនៃ amyotrophy ឆ្អឹងខ្នង។
មិនមានការព្យាបាលជាក់លាក់សម្រាប់ Kugelberg-Welander amyotrophy ទេ។ ភ្នាក់ងាររោគសញ្ញានិងស្តារឡើងវិញត្រូវបានប្រើ។ ជម្រើសត្រឹមត្រូវនៃវិជ្ជាជីវៈ និងការលុបបំបាត់ការផ្ទុកលើសទម្ងន់រាងកាយមានសារៈសំខាន់។
រោគសញ្ញា glenohumeral-facial សរសៃប្រសាទ (បំរែបំរួលឆ្អឹងខ្នងនៃជំងឺ myopathy Landouzi-Dejerine)
ក្នុងករណីខ្លះជាមួយនឹង amyotrophy ឆ្អឹងខ្នង ការធ្វើមូលដ្ឋានីយកម្មនៃ atrophies គឺជាលក្ខណៈនៃ Landouzi-Dejerine myodystrophy ពោលគឺវាទាក់ទងនឹងសាច់ដុំនៃក្រវ៉ាត់ស្មាជាពិសេស scapulae ជួសជុល ផ្នែកជិតនៃអវយវៈខាងលើ (សាច់ដុំ biceps និង triceps brachii) ។ និងសាច់ដុំមុខ។ ការសិក្សាអំពីអេឡិចត្រូម៉ាញេទិកបង្ហាញពីទំហំខ្ពស់ កាត់បន្ថយសកម្មភាពជីវអគ្គិសនី ជាមួយនឹងសក្ដានុពលនៃការបំភាន់ច្បាស់លាស់ ពោលគឺរូបភាពលក្ខណៈនៃកម្រិតឆ្អឹងខ្នងនៃដំបៅ។ សកម្មភាពនៃអង់ស៊ីមនៅក្នុងសេរ៉ូមឈាមចំពោះអ្នកជំងឺបែបនេះជាធម្មតាមានលក្ខណៈធម្មតាសូចនាករនៃការរំលាយអាហារ creatine-creatinine ស្ទើរតែមិនផ្លាស់ប្តូរ។ បច្ចុប្បន្ននេះ ការពិពណ៌នាជាច្រើននៃករណីស្រដៀងគ្នានេះ បានប្រមូលផ្តុំនៅក្នុងអក្សរសិល្ប៍ ហើយអ្នកនិពន្ធមួយចំនួនកំណត់អត្តសញ្ញាណជំងឺសាច់ដុំ neurogenic atrophy ដែលនឹកឃើញដល់ទម្រង់ Landouzy-Dejerine ។ការចាប់ផ្តើមនៃជំងឺនេះដូចទៅនឹង Landouzy-Dejerine myodystrophy កើតឡើងនៅអាយុផ្សេងៗគ្នា - ទាំងក្នុងវ័យកុមារភាពនិងក្នុងវ័យពេញវ័យ (ពី 7 ទៅ 40 ឆ្នាំ) ។ ជំងឺនេះមានដំណើរការធម្មតា ហើយការវិវត្តន៍យឺត។ នៅក្នុងវ៉ារ្យ៉ង់ឆ្អឹងខ្នងនៃជំងឺ Landouzi-Dejerine ភាពមិនស្មើគ្នានៃដំបៅត្រូវបានបង្ហាញកាន់តែច្បាស់។ ការផ្លាស់ប្តូរបេះដូងដែលត្រូវបានកត់ត្រាដោយភាពមិនប្រក្រតីនៃ ECG គឺជារឿងធម្មតាដែលផ្ទុយទៅនឹងជំងឺសាច់ដុំ glenohumeral ។ ការខូចខាតដល់សាច់ដុំមុខអាចមានតិចតួច ឬត្រូវបានរកឃើញយឺត។
អ្នកនិពន្ធខ្លះចាត់ទុកទម្រង់ scapular-peroneal នៃ amyotrophy ជាប្រភេទនៃការប្រែប្រួល neurogenic នៃ Landouzi-Dejerine myodystrophy ។ នៅក្នុងករណីទាំងនេះ, ការចូលរួមរបស់បេះដូងនៅក្នុងដំណើរការ pathological ពេលខ្លះត្រូវបានពិពណ៌នា។
ទម្រង់ដ៏កម្រនៃសាច់ដុំឆ្អឹងខ្នង
ទម្រង់ដ៏កម្រនៃ amyotrophy ឆ្អឹងខ្នងរួមមានការដាច់សាច់ដុំដាច់ពីគ្នា។ ជំងឺនេះចាប់ផ្តើមនៅផ្នែកចុងនៃអវយវៈទាប ផ្នែកចុងនៃដៃត្រូវបានចូលរួមជាបណ្តើរៗក្នុងដំណើរការ ហើយការទូទៅនៃដំណើរការអាចត្រូវបានគេសង្កេតឃើញ។ទម្រង់ neurogenic នៃ oculopharyngeal atrophy ដែលត្រូវបានបញ្ជូនតាមលក្ខណៈ autosomal dominant ត្រូវបានពិពណ៌នា។ អ្នកនិពន្ធបានរាយការណ៍ករណីមួយដែលការធ្វើកោសល្យវិច័យបានបង្ហាញពីការចុះខ្សោយនៃកោសិកានៅក្នុងស្នែងផ្នែកខាងមុខនៃខួរឆ្អឹងខ្នងនិងស្នូលម៉ូទ័រនៃសរសៃប្រសាទ cranial រួមទាំង nuclei III និង X គូ។
amyotrophies ឆ្អឹងខ្នងរួមមានករណីភាគច្រើននៃជម្ងឺ arthrogrypposis ពីកំណើត។ ដំណើរការ pathological រួមមានការវិវត្តន៍នៃកោសិកានៃស្នែងផ្នែកខាងមុខនៃខួរឆ្អឹងខ្នងជាមួយនឹង paresis នៃសាច់ដុំដែលត្រូវគ្នា។ ជាលទ្ធផលនៃការទាញសាច់ដុំមិនស្មើគ្នានៅក្នុងស្បូន ការកន្ត្រាក់ និងការវិវត្តន៍មិនធម្មតានៃសន្លាក់អាចបង្កើតបាន។ ក្នុងអំឡុងពេលធ្វើកោសល្យវិច័យនិងការសិក្សា EMG ក្នុងករណីខ្លះការផ្លាស់ប្តូរ neurogenic និង myogenic ត្រូវបានកត់សម្គាល់ហើយដូច្នេះពាក្យ "pseudomyopathy" ត្រូវបានស្នើឡើង។
វាក៏មានទម្រង់មិនខុសគ្នានៃ amyotrophy ឆ្អឹងខ្នងជាមួយនឹងវគ្គសិក្សាដែលរីកចម្រើនយ៉ាងឆាប់រហ័ស រីកចម្រើនយឺត និងមិនរីកចម្រើន។
អាមីតូត្រូហ្វី - ការរំលោភលើសាច់ដុំ trophism ដោយសារតែការខូចខាតដល់សរសៃប្រសាទម៉ូទ័រគ្រឿងកុំព្យូទ័រ អមដោយការផ្លាស់ប្តូរ degenerative-dystrophic ភាពស្តើង និងមុខងារ contractile ចុះខ្សោយ។ ក្នុងចំណោមជំងឺឆ្អឹងខ្នង A., ជំងឺ Werdnig-Hoffmann, ទម្រង់រីកចម្រើន pseudomyopathic នៃ Kugelberg-Welander, ជំងឺ Aran-Duchenne ត្រូវបានសម្គាល់ ការបង្ហាញគ្លីនិកគឺការវិវឌ្ឍន៍បន្តិចម្តងៗនៃភាពខ្វិនខ្វិន និងសាច់ដុំ atrophy, asymmetry នៃដំបៅ និងអវត្តមាននៃការឆ្លុះសរសៃពួរ។ . ភាពរសើប និងមុខងារនៃសរីរាង្គអាងត្រគាកជាធម្មតាមិនចុះខ្សោយទេ។ ប្រតិកម្មគុណភាពនៃ degeneration ត្រូវបានកត់សម្គាល់នៅពេលសិក្សាពីភាពរំជើបរំជួលអគ្គិសនី។ អាមីតូត្រូហ្វីតឆ្អឹងខ្នង Werdnig-Hoffmann . ជំងឺ autosomal recessive ។ បែងចែក ពីកំណើត កុមារភាពដំបូង និងទម្រង់យឺតនៃជំងឺ។ទម្រង់ពីកំណើតអាចបង្ហាញខ្លួនវានៅក្នុងអំឡុងពេលសម្រាល (ចលនាគភ៌យឺត និងយឺតត្រូវបានកត់សម្គាល់) ឬក្នុងខែដំបូងនៃជីវិតរបស់កុមារ។ កុមារកើតមកមាន paresis ខ្សោយ សាច់ដុំ hypotonia ទូទៅ និងអវត្តមាននៃការឆ្លុះសរសៃពួរ។ មានតែកុមារពីរបីនាក់ប៉ុណ្ណោះដែលអភិវឌ្ឍសមត្ថភាពក្នុងការលើកក្បាលរបស់ពួកគេឡើង និងអង្គុយដោយឯករាជ្យ។ វគ្គសិក្សាកំពុងរីកចម្រើនយ៉ាងឆាប់រហ័ស។ មូលហេតុចម្បងនៃការស្លាប់គឺការបរាជ័យផ្លូវដង្ហើម និងសរសៃឈាមបេះដូង ដោយសារភាពទន់ខ្សោយនៃសាច់ដុំផ្លូវដង្ហើម និងជំងឺ bulbar ។
ទម្រង់យឺតចាប់ផ្តើមបន្តិចម្តង ៗ នៅអាយុ 1.5-2.5 ឆ្នាំ។ ចលនា និងការដើររបស់កុមារមានភាពមិនច្បាស់លាស់ ហើយជារឿយៗកុមារដួល paresis flaccid និង atrophy នៃសាច់ដុំនៃអវយវៈជិតលេចឡើង។ ការឆ្លុះបញ្ចាំង Tendon ត្រូវបានកាត់បន្ថយ។ សាច់ដុំ hypotonia រួមចំណែកដល់ការវិវត្តនៃការខូចទ្រង់ទ្រាយនៃទ្រូងនិងភាពធូររលុងនៃសន្លាក់។ ការឡើងរឹងនៃសាច់ដុំអណ្តាត និងការថយចុះនៃការឆ្លុះនៃ pharyngeal និង palatal គឺជារឿងធម្មតា។ រោគសញ្ញា Bulbar ជាមួយនឹង dysphagia មានការរីកចម្រើនបន្តិចម្តង ៗ ជាមួយនឹងទម្រង់ A. អ្នកជំងឺរស់នៅរហូតដល់ 20-30 ឆ្នាំ។
ការធ្វើរោគវិនិច្ឆ័យត្រូវបានធ្វើឡើងដោយផ្អែកលើទិន្នន័យគ្លីនិក។
ការព្យាបាល. ការព្យាបាលដោយលំហាត់ប្រាណ ម៉ាស្សា និងថ្នាំដែលធ្វើអោយប្រសើរឡើងនូវ trophism នៃជាលិកាសរសៃប្រសាទត្រូវបានចេញវេជ្ជបញ្ជា - cerebrolysin, aminalon, pyriditol ។
Amyotrophy ឆ្អឹងខ្នង Kugelberg - Welander ជំងឺ autosomal recessive ឬ autosomal dominant disease ចាប់ផ្តើមនៅក្នុងករណីភាគច្រើននៅក្នុង អាយុ 4-8 ឆ្នាំ, ពេលខ្លះក្រោយមក។អស់កម្លាំង ភាពទន់ខ្សោយទូទៅ ភាពទន់ខ្សោយនៅក្នុងជើង (ជាពិសេសនៅពេលឡើងជណ្តើរ) និងការរមួលសាច់ដុំលេចឡើង។ សាច់ដុំ atrophy មានការរីកចម្រើនបន្តិចម្តងៗ ដែលអាចត្រូវបានបិទបាំងដោយស្រទាប់ខ្លាញ់ subcutaneous ។ ការផ្លាស់ប្តូរការដើរ សម្លេងសាច់ដុំថយចុះ ការឆ្លុះសរសៃពួរបាត់ ហើយជួរនៃចលនាសកម្មថយចុះ (flaccid paresis)។ នៅលើការពិនិត្យ, អ្វីដែលគេហៅថា pseudohypertrophy នៃសាច់ដុំកំភួនជើងត្រូវបានកត់សម្គាល់ (ការកើនឡើងនៃបរិមាណរបស់ពួកគេដោយសារតែការវិវត្តនៃជាលិកា adipose) ពីរបីឆ្នាំបន្ទាប់ពីការបង្ហាញនៃ A. នៅក្នុងចុងទាប, atrophy និង fascicular រមួលលេចឡើងនៅក្នុង។ ក្រុមសាច់ដុំជិតនៃចុងខាងលើ (ប្រភេទ A.) ។ វគ្គសិក្សាមានការរីកចម្រើនបន្តិចម្តង ៗ សកម្មភាពម៉ូទ័រនៅតែបន្តកើតមានយូរ។ អ្នកជំងឺរស់នៅរហូតដល់ 40-50 ឆ្នាំជាញឹកញាប់មានសមត្ថភាពក្នុងការថែទាំខ្លួនឯង។ នៅពេលដែលរោគសញ្ញា bulbar លេចឡើងក្នុងដំណាក់កាលក្រោយ ការព្យាករណ៍កាន់តែអាក្រក់ទៅ ៗ ។
រោគវិនិច្ឆ័យដោយផ្អែកលើគ្លីនីកលទ្ធផលនៃ electromyography សកលនិងម្ជុលនិងការសិក្សា morphological នៃសាច់ដុំគ្រោងឆ្អឹងដែលអនុញ្ញាតឱ្យយើងកំណត់អត្តសញ្ញាណធម្មជាតិ denervation នៃការផ្លាស់ប្តូរ។
អាមីតូត្រូហ្វីតឆ្អឹងខ្នងសម្រាប់មនុស្សពេញវ័យ (ជំងឺ Aran-Duchenne) . ជាកម្មសិទ្ធិរបស់ជំងឺនេះចំពោះឆ្អឹងខ្នង A. មិនត្រូវបានទទួលស្គាល់ដោយអ្នកស្រាវជ្រាវទាំងអស់ទេ។ ជំងឺនេះចាប់ផ្តើមនៅអាយុ 40-60 ឆ្នាំ។ ការចុះខ្សោយនៃសាច់ដុំនៃអវយវៈចុង (ជាធម្មតាដៃ) មានការរីកចម្រើនបន្តិចម្តងៗ។ ក្រោយមកទៀត សាច់ដុំនៃអវយវៈជិតៗ អាងត្រគាក និងក្រវាត់ស្មា ក៏ចូលរួមក្នុងដំណើរការនេះដែរ។ មាន fasciculations នៅក្នុងសាច់ដុំដែលរងផលប៉ះពាល់, និងការ fibrillations នៅក្នុងសាច់ដុំនៃអណ្តាត។ វគ្គសិក្សាកំពុងរីកចម្រើនបន្តិចម្តង ៗ ។ ការស្លាប់ជាធម្មតាកើតឡើងដោយសារជំងឺរលាកទងសួត។